
在迷雾中前行的AAV疗法
更新时间:
2025-07-22 08:26:21.756

基因治疗是指通过向患者体内引入特定的细胞功能改变遗传物质来治疗遗传疾病的方法。实现方式包括体外或体内实施两种:体外基因治疗涉及提取患者的细胞,对其进行基因改造,然后重新注入患者体内;体内基因治疗则是直接将遗传物质递送到目标组织。
基因治疗的关键在于将基因高效地递送到目标组织或细胞中,这一过程由称为载体的基因递送工具完成。载体分为病毒性和非病毒性两种,目前,共有43种基因治疗获批(图1),其中,17种采用病毒载体(包括AAV、腺病毒、逆转录病毒、慢病毒和单纯疱疹病毒),而26种采用非病毒载体(包括反义寡核苷酸、siRNA以及基于细胞的CRISPR基因编辑技术)。
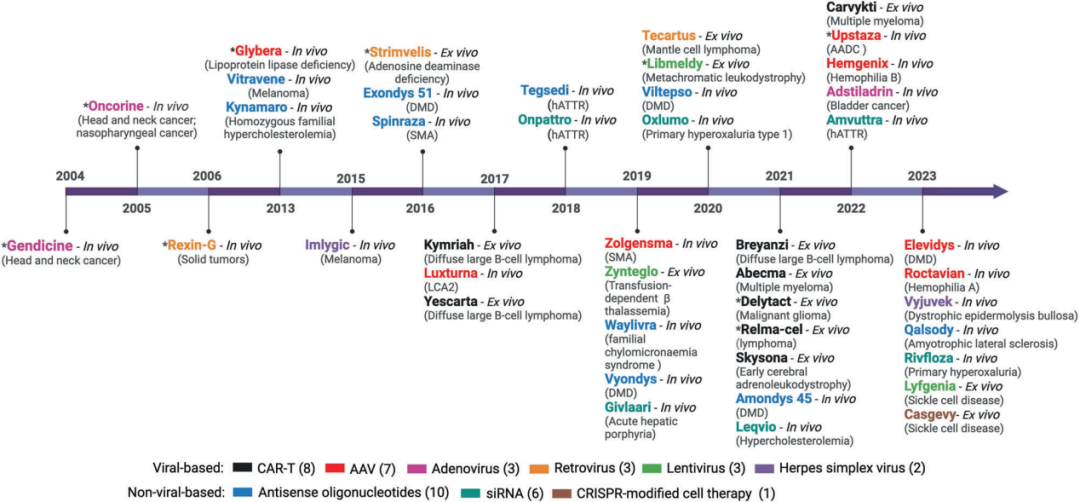
图1 获批的基因治疗产品和递送平台
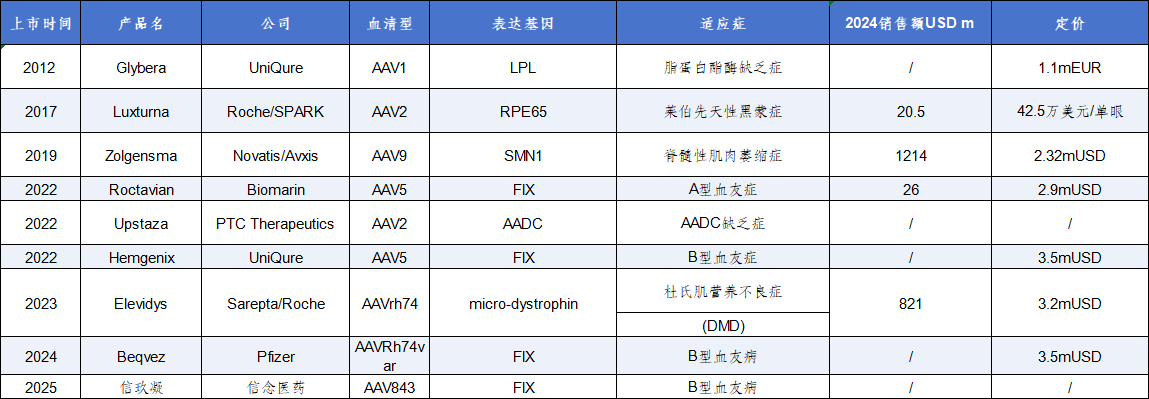
在in vivo基因治疗中,腺相关病毒(AAV)载体因其广泛的组织亲和性、相对良好的安全性以及多样的生产方式而受到开发者青睐,目前已有9款AAV基因治疗产品获批上市(表1)。
表1 已获批AAV基因治疗产品
TONACEA
AAV疗法基本原理
腺相关病毒(Adeno-associated virus,AAV)是目前发现结构最简单的、无包膜的单链DNA病毒,由蛋白衣壳(capside)和长度为4.7kb的单链DNA基因组构成,衣壳长度为20 ~ 25nm,属于细小病毒家族。AAV自身不能复制,必须依赖于其它病毒复制,比如腺病毒、疱疹病毒、杆状病毒,有超过80%的人携带AAV,未发现AAV与任何疾病相关。
病毒(Recombinant AAV,rAAV)是经工程改造的AAV载体,它剔除了AAV基因组全部编码蛋白Rep或Cap基因序列,只在两端保留作为包装信号的反式DNA序列(即ITR)。用于基因疗法载体的重组腺相关病毒(rAAV)携带的蛋白衣壳与野生型AAV几乎完全相同,然而基因组中编码病毒蛋白的部分完全被治疗性转基因(transgene)替代。
AAV基因疗法应用策略主要有三类:
1、基因置换:最常见的AAV介导的治疗干预是基因置换,其特点是引入功能基因副本治疗单基因疾病。该方法主要用于罕见和不可治疗的疾病,如莱伯氏先天性黑蒙症(一种遗传性视网膜疾病,Luxturna于2017年获得批准用于治疗该疾病)、小儿脊髓性肌萎缩症,(Zolgensma于2019年获得批准用于治疗该疾病)以及血友病A(凝血蛋白因子VIII缺乏引起,一种隐性x连锁疾病,Roctavian余2022年获得批准用于治疗该疾病)。
2、基因添加:基因添加是AAV基因治疗应用最广泛的一种,因为它可以用于治疗较常见的非单基因复杂疾病,如慢性、自身免疫性或感染性疾病。使用AAV进行基因添加的例子包括正在进行的治疗类风湿性关节炎的临床试验。目前,Arthrogen正在进行1期临床试验(NCT02727764)。AAV载体携带IFN-β转基因,IFN-β是一种抗炎介质,在骨骼和软骨保护中发挥作用。
3、基因沉默:与试图克服功能缺失突变的基因替换不同,基因沉默,也被称为RNA干扰(RNAi),可以用来抑制有害的功能获得突变。在这种方法中,AAV转基因编码一个miRNA前体,该前体经靶细胞自身的RNAi机制加工产生miRNA,这是一个小的非编码RNA链,可以与mRNA对中的互补序列建立碱基,以抑制其表达。利用AAV进行基因沉默的大多数努力仍处于临床前研究阶段,因为沉默有几个必须解决的具体安全问题。一个主要问题是非目标沉默的可能性。另一个问题是RNAi饱和的机制。
在这个机制中,转基因miRNA的表达可能压倒并破坏内源性miRNA的产生,导致细胞毒性。目前,unique已经开始了一项使用AAV基因疗法治疗亨廷顿舞蹈病的临床试验(NCT049493),需要在核磁共振(MRI)成像的引导下,将该药物精准注射到大脑的壳核和尾状核区域。
TONACEA
罕见病患者的救命稻草
AAV疗法作为最先进的治疗方法之一,给无药可治的罕见病患者带来了新的希望。
1
AAV疗法与血友病
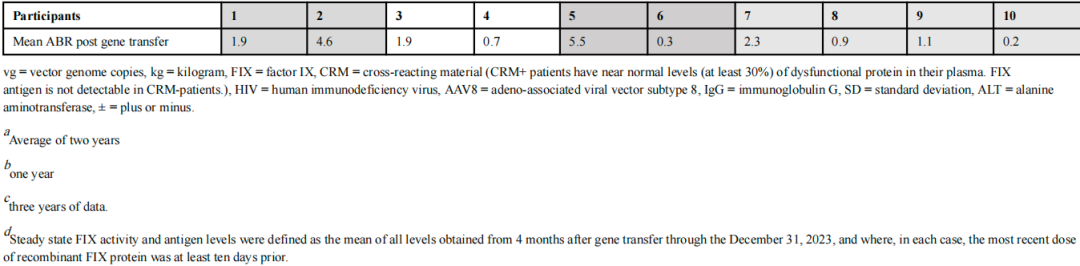
2025年7月4日,NEJM发表了10例重度血友病B患者接受scAAV2/8-LP1-hFIXco单次快速输注治疗随访13年的结果(NCT00979238):
除短暂性肝转氨酶升高外,未观察到长期或新的AAV相关不良事件。因子IX表达保持稳定,十名参与者中有七名未服用因子IX预防药物。临床上,AAV基因转移使ABR和因子IX浓缩物用量减少了9倍以上,显著减轻了疾病负担。
两名受试者出现了肿瘤病变,根据研究方案,这些病变被报告为可能与AAV相关的严重不良事件。然而,后续的分子研究和专家多学科评审表明,这些事件可能与AAV基因治疗无关,而是像之前描述的一样,归因于普通人群中普遍存在的年龄相关或环境风险因素。
表2 10名患者基线及接受治疗后特征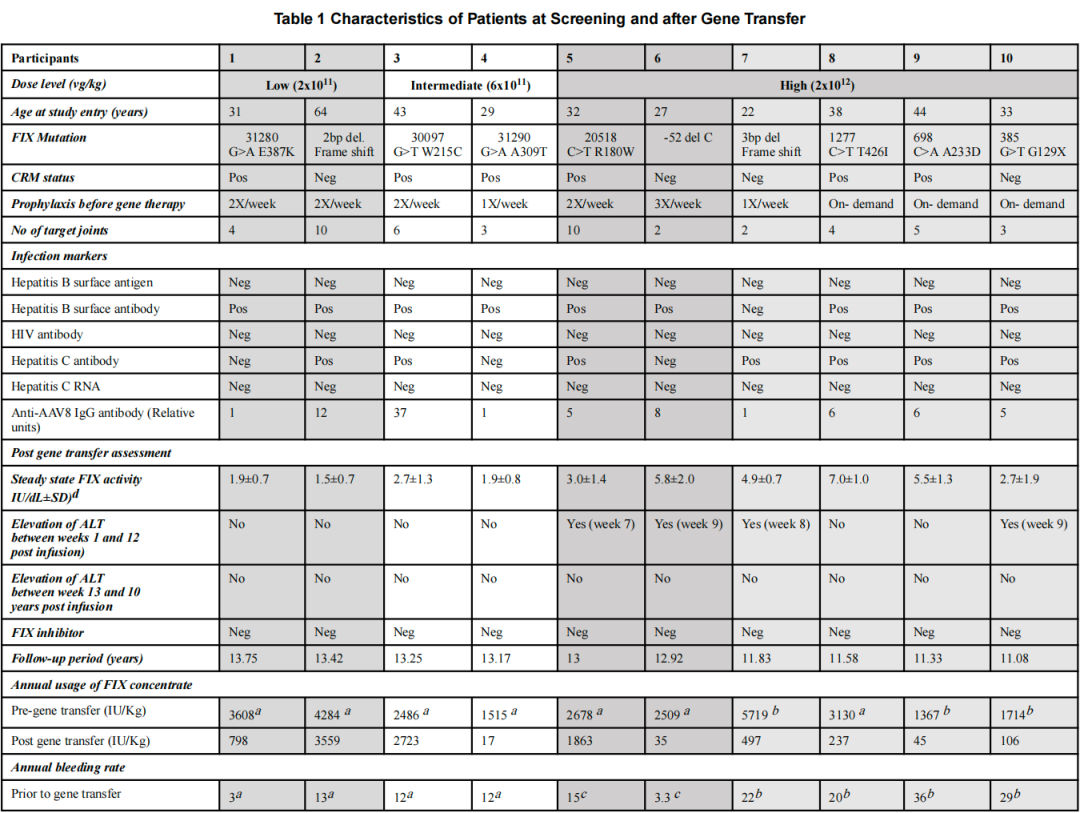
2
AAV疗法与莱伯先天性黑蒙症
莱伯先天性黑蒙症是一组遗传性视网膜疾病,会导致婴儿期视力严重下降。莱伯先天性黑蒙症的发病机制涉及20多个基因,这些基因最常参与光感受器纤毛运输、光传导、视觉周期和光感受器发育。
首个针对遗传性视网膜疾病的基因疗法voretigene neparvovec-rzyl(Luxturna,Spark Therapeutics)于2017年获批,用于治疗由RPE65突变引起的莱伯先天性黑蒙。这种视网膜下基因疗法改善了功能性视力,并且大多数患者耐受性良好。从那时起,几种视网膜基因疗法一直在开发和临床试验中。
ATSN-101是针对LCA1的基因疗法。2024年9月,Atsena发表了从ATSN-101的1/2期试验到治疗后12个月的所有患者的安全性和有效性数据:
2019年9月12日至2022年5月5日期间,15名经基因确诊为LCA1的患者参与了本研究,所有患者均于2023年6月前完成了第12个月的随访。
15名患者中,有13名(87%)接受了足量的300 μL视网膜下注射(包括高剂量组的所有9名患者);队列1和2中各有一名患者的视网膜下注射量略有减少,分别约为200 μL和250 μL。
对于高剂量组,合并后,治疗和未治疗的眼睛在白色和彩色刺激方面均存在显着差异。在第28天观察到高剂量治疗的眼睛相对于未治疗的眼睛有所改善,并且这种改善持续到第12个月;治疗的眼睛在第12个月改善了20.3 dB,而未治疗的眼睛仅改善了1.1dB。接受高剂量治疗的9名患者中,有6名在暗适应白色FST中表现出超过10 dB的改善,改善幅度高达46.5 dB。
治疗12个月后,所有13名完成色觉刺激暗适应测试的患者均表现出视杆介导的反应,蓝色和红色敏感度之间约有2个对数单位的差异(21.7至28.7 dB);除一名表现出视锥介导反应的患者外,所有患者的基线反应类似地都是视杆介导的。与暗适应FST相反,在光适应FST中没有观察到一致的改善;在11名完成色觉刺激光适应测试的患者中,只有两名在治疗12个月后表现出视锥介导的反应,而这些患者在基线时都有视锥介导的反应。
3
AAV疗法与常染色体隐性遗传性耳聋9型(DFNB9)
OTOF基因突变是导致常染色体隐性遗传性耳聋9型(DFNB9)的核心病因,患者因内耳毛细胞突触囊泡功能缺陷而丧失听力。人工耳蜗是重度DFNB9患者的主要治疗方法,虽能部分恢复听觉,但存在音质失真、噪声环境识别困难等局限。OTOF基因疗法则直击疾病根源——通过腺相关病毒(AAV)载体将功能性OTOF基因精准递送至耳蜗内毛细胞,修复突触传递功能。
2025年7月2日,《Nature Medicine》发表了中国研究团队在中国的五个医学中心对10名年龄在1.5-23.9岁之间的常染色体隐性遗传耳聋9型(DFNB9)患者进行的一项使用Anc80L65衣壳的AAV-OTOF基因治疗的单臂临床试验。临床试验的主要终点指标为5年内的安全性和耐受性,次要终点指标评估听觉功能。
对10名随访了6-12个月的患者(其中包括1名接受了两次注射治疗的患者)的初步研究结果表明,该AAV-OTOF基因疗法耐受性和安全性良好,共出现162起1/2级不良事件,中性粒细胞百分比降低是最常见的不良事件(在162例中有16例)。
所有10名患者均接受了至少6个月的随访,他们的平均纯音听阈从基线的106分贝改善至52分贝(正常谈话时的音量水平)。其他次要终点指标也显示出类似改善,包括平均点击声听觉脑干反应(ABR)阈值、短纯音ABR阈值和听觉稳态反应(分别从101分贝改善至48分贝、91分贝改善至57分贝、80分贝改善至64分贝)。
TONACEA
肝毒性阴影笼罩
AAV的肝毒性引起的患者死亡事件时见公布,引起监管警惕与业内担忧:
• 2025年3月,Sarepta公司报告称,一名16岁患者在接受其已获批上市的杜氏肌营养不良症基因疗法Elevidys治疗后发生了严重急性肝损伤并最终导致死亡。2025年6月15日,Sarepta再度发布公告,表示Elevidys出现第二例由ALF导致的死亡病例。而后FDA发布的公告中明确指出,目前已确定两例死亡病例均与Elevidys治疗存在关联,患者均在接受治疗后两个月内出现转氨酶升高并住院,最终进展为ALF而死亡。之后将进一步评估Elevidys引发ALF(导致住院或死亡等严重后果)的风险,并考虑是否需采取进一步监管措施(如更新黑框警告、限制使用人群或撤市)。
• 2022年,诺华(Novartis)报告了两名患儿在接受用于治疗脊髓性肌萎缩症的Zolgensma治疗后,因急性肝功能衰竭而发生的死亡。
• 2020年至2021年间,安斯泰来研发的用于治疗X连锁肌管病(XLMTM)的AAV8基因疗法AT132,在其ASPIRO临床研究(NCTO3199469)的高剂量组(3 x 10^14 vg/kg)中,先后导致4名患儿因严重的免疫介导肝损伤、肝功能衰竭及继发性脓毒症死亡。而后该项目彻底终止。
TONACEA
明暗难辨的前路
聚焦罕见病事实上对AAV疗法的开发者来说是把双刃剑,一方面,罕见病背后的各种快速注册制度允许开发者通过少量患者的临床数据实现快速上市,另一方面,狭窄的受众人群,给商业化带来了难以逾越的障碍,即使强大如pfizer也不得不壮士断腕。
• 2025年3月,罗氏对Spark启动“根本性重组”,计提24亿美元商誉减值,裁撤337个岗位并将剩余310名员工并入母公司,Spark首款获批基因疗法产品Luxturna(治疗遗传性视网膜疾病疗法)2023年销售额暴跌59%,从2022年的5000万美元暴跌至约2000万美元。罗氏直言Spark的未来收入与协同效应无法覆盖账面价值,因此采取重组。
• 2025年2月27日,Vertex(福泰制药)终止与Verve Therapeutics价值4.66亿美元的肝病基因编辑合作,收回2022年以来投入的2500万美元首付款及3500万美元股权投资。
• 2025年,辉瑞(Pfizer)因零患者使用宣布Beqvez撤市,清空了基因疗法的商业和临床管线,此前,其DMD基因疗法fordadistrogene movaparvovecIII期临床失败,与Sangamo合作的A型血友病项目giroctocogene fitelparvovec亦于2024年末终止。
• 2023年武田制药(Takeda)宣布结束早期的AAV项目,相关项目成员成立了Crosswalk,并收购了相关产品
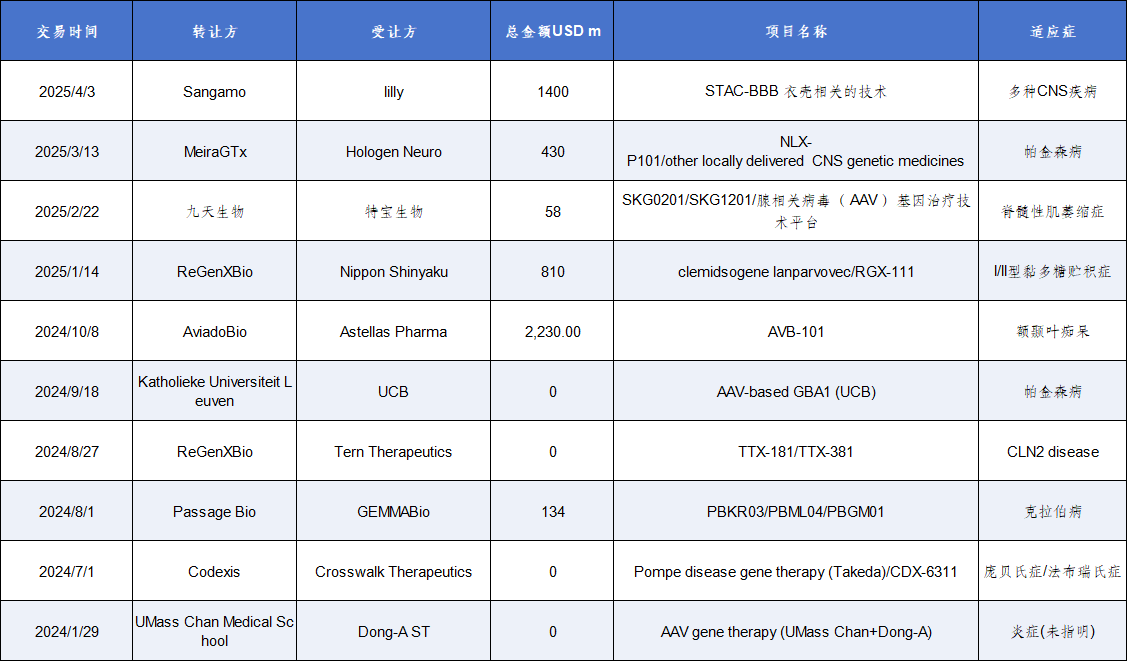
但与此同时,依然有玩家看好AAV的前景,尤其是在CNS领域的潜力,因此衍生的BD交易也并不鲜见。
2025年Sangamo与礼来达成协议,将获得Sangamo专有的嗜神经性(neurotropic)腺相关病毒(AAV)衣壳STAC-BBB在一个初始疾病靶点上的全球独家使用权,还可以增加最多四个神经系统疾病靶点,以开发针对中枢神经系统(CNS)特定疾病的基因疗法。Sangamo将从礼来获得1800万美元的预付款,并将在五个潜在的靶点上获得高达14亿美元的额外靶点费和里程碑付款,以及潜在净销售额的分层特许权使用费。
表3 AAV疗法的BD交易
— 结语 —
AAV疗法作为基因疗法的成功范本给许多无药可治的罕见病患者带来了康复的希望,但不断出现的肝毒性事件却为此笼罩上了一层阴影,与此同时,艰难的商业化环境进一步恶化了基因疗法biotech的生存环境,让AAV的未来显得明暗难辨,但也请期待新的改造技术的出现,实现疗效、安全性与商业化的三赢。
参考文献:(上下滑动查看更多)
1.Wang H, Chen Y, Lv J, Cheng X, Cao Q, Wang D, Zhang L, Zhu B, Shen M, Xu C, Xun M, Wang Z, Tang H, Hu S, Cui C, Jiang L, Yin Y, Guo L, Zhou Y, Han L, Gao Z, Zhang J, Yu S, Gao K, Wang J, Chen B, Wang W, Chen ZY, Li H, Shu Y. Bilateral gene therapy in children with autosomal recessive deafness 9: single-arm trial results. Nat Med. 2024 Jul;30(7):1898-1904. doi: 10.1038/s41591-024-03023-5. Epub 2024 Jun 5. PMID: 38839897; PMCID: PMC11271389.
2.Reiss UM, Davidoff AM, Tuddenham EGD, Chowdary P, McIntosh J, Muczynski V, Journou M, Simini G, Ireland L, Mohamed S, Riddell A, Pie AJ, Hall A, Quaglia A, Mangles S, Mahlangu J, Haley K, Recht M, Shen YM, Halka KG, Fortner G, Morton CL, Gu Z, Hayden RT, Neufeld EJ, Okhomina VI, Kang G, Nathwani AC. Sustained Clinical Benefit of AAV Gene Therapy in Severe Hemophilia B. N Engl J Med. 2025 Jun 12;392(22):2226-2234. doi: 10.1056/NEJMoa2414783. PMID: 40499172; PMCID: PMC7617823.
3.Yang P, Pardon LP, Ho AC, Lauer AK, Yoon D, Boye SE, Boye SL, Roman AJ, Wu V, Garafalo AV, Sumaroka A, Swider M, Viarbitskaya I, Aleman TS, Pennesi ME, Kay CN, Fujita KP, Cideciyan AV. Safety and efficacy of ATSN-101 in patients with Leber congenital amaurosis caused by biallelic mutations in GUCY2D: a phase 1/2, multicentre, open-label, unilateral dose escalation study. Lancet. 2024 Sep 7;404(10456):962-970. doi: 10.1016/S0140-6736(24)01447-8. PMID: 39244273.
4.Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019 May;18(5):358-378. doi: 10.1038/s41573-019-0012-9. PMID: 30710128; PMCID: PMC6927556.
5.Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther. 2021 Feb 8;6(1):53. doi: 10.1038/s41392-021-00487-6. PMID: 33558455; PMCID: PMC7868676.
上一页
下一页
相关新闻

联系我们
同写意

写意宣发

同写意Biotech

同写意微服务

©2022 同写意(北京)科技发展有限公司