
Drug RA丨第16章 药品临床1-4期-中东北非地区(埃及、黎巴嫩、沙特阿拉伯王国)及非洲(南非)
更新时间:
2025-08-15 08:14:55.825
基于上3篇:第16章 药品临床1-4期-美洲法规要求(美国、阿根廷、加拿大、智利、墨西哥)
第16章 药品临床1-4期-欧洲法规要求(欧盟、瑞士、英国)
第16章 药品临床1-4期-金砖四国(巴西、俄罗斯、印度、中国)和亚太地区(澳大利亚、日本、新西兰、新加坡)
现介绍中东/北非地区(埃及、黎巴嫩、沙特阿拉伯王国)及非洲临床试验法规。
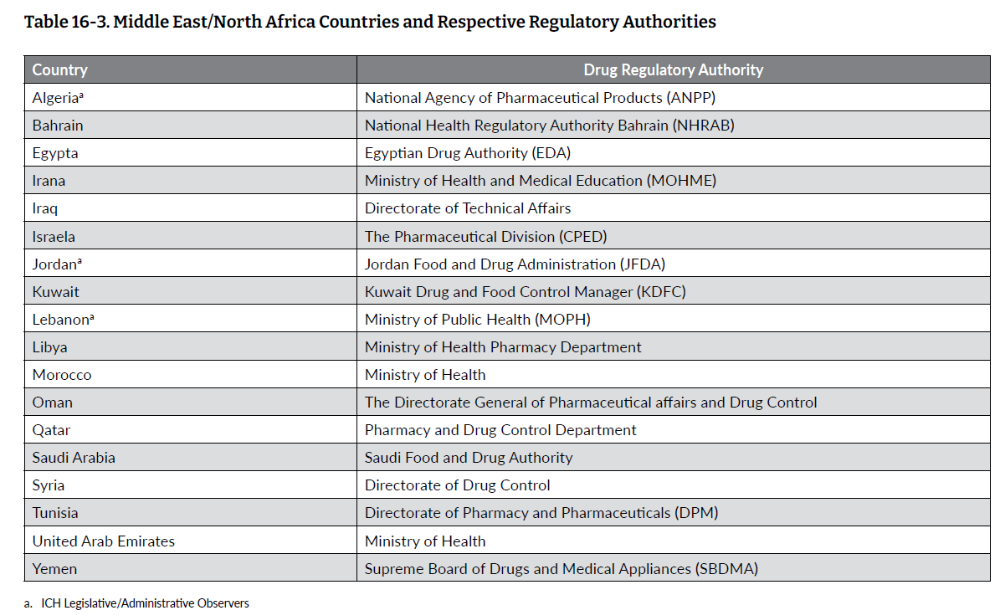
中东和北非地区一般被认为包括非洲大陆最北端和亚洲西南部介于地中海和波斯湾之间的国家,最西端是摩洛哥,向东延伸至伊朗。该地区约有 4.2 亿人口,人口最多的国家是埃及,有 1.03 亿人,其次是伊朗,有 8500 万人。该地区有几个国家是 ICH 观察员(表 16-3),大多数国家都通过立法建立了药品监管机构,但在这些地区,各国的药品监管实践差异很大,而且经常处于发展阶段,因此申办者很难知道在某一特定时间点实施了哪些法规。
除埃及外,这些地区的临床试验很少。随着人口不断增长,对新型疗法的需求也在不断增加。中东和北非地区缺乏临床研究基础设施,研究参与者对临床试验的基本原理缺乏了解,这些都是该地区临床试验发展的主要障碍。
在许多中东和北非国家,治疗产品(包括药物和生物制品)临床试验的监管实践仍在进行中,申办者在该地区开展试验工作时,必须认真关注不断发展的法律、政策和实践。
• 药品监管协调现状
虽然中东和北非地区有多个 ICH 观察员/成员国(表 16-3),但中东和北非国家在药物开发方面并未统一或一致地采用ICH,包括 ICHE6(R2) GCP 指南。该地区的一些国家已尝试协调药品监管实践。由巴林、阿曼、沙特阿拉伯、科威特、阿联酋和卡塔尔组成的海湾合作委员会(GCC)于 2014 年成立,并设立了药物中央注册机构,该机构的目标是根据 ICH 指南,在治疗产品质量、安全性和有效性等关键要素方面协调成员国的药物审批实践。
本节将重点介绍埃及、黎巴嫩和沙特阿拉伯王国的临床试验审批和监管情况。
TONACEA
埃及
埃及的药品监管机构是埃及药品管理局(Egyptian Drug Authority,EDA),它是埃及卫生部 (MoH) 下属的一个政府实体。EDA 在埃及是一个相对较新的实体,是根据第 151/2019 号法律成立的,该法律旨在取代埃及早期的 1955 年第 127 号法律。
该法于 2019 年底生效,以取代埃及原有的药品监管框架,该框架由卫生部内的三个独立实体(药品事务中央管理局、国家药品控制和研究组织以及国家生物制品研究和控制组织)管理。根据 2019 年第 151 号法律和 EDA,埃及着手简化其治疗开发监督、注册和上市后监督的方法,包括使埃及的监管实践更加符合 ICH 准则的原则。该法律还旨在帮助刺激埃及的药品生产。
埃及药品管理局下设三个行政实体,负责管理埃及药品管理局的临床试验法规: 临床试验评估管理部门、临床方案和后续行动管理部门以及科学委员会和技术支持管理部门。这三个实体隶属于 EDA 的生物和创新产品及临床研究中央管理局 (BIO-INN)。BIO-INN 及其行政部门的法定职权范围来自埃及临床试验法(第 214/2020 号法律)。该法也被称为 “临床研究法”,是埃及第一部此类法律。
向 EDA 提交新的临床研究以获得继续进行的授权为分两步的连续过程,在向 BIO-INN 提交之前,必须首先获得 IRB 批准。BIO-INN 就 CTA 所需的内容提供详细指导、其中包括但不限于:
填写完整的 “申请人向埃及药品管理局提交的人用医药产品临床试验授权申请”。
• IRB 批准
• 临床研究方案
• 知情同意书
• IB
• 与所有参与的埃及临床研究机构签订 CTA
• 病例报告表
对于 FIH 1 期试验,EDA 制定了单独的指南,其中概述了支持此类早期阶段研究所需的非临床数据:临床前要求清单:“研究者和/或申办者/CRO 档案中临床前所需文件清单,在首次用于人体之前提交 EDA 以征求科学意见 ”。这些要求与支持 FIH 研究所需的非临床研究的 ICH 指南一致。
对于所有提交的 CTA,EDA 表示大约需要 60 个工作日来完成审查和批准过程,其中包括解决在此过程中发送给申办者的询问。
埃及药品管理局明确表示,已完全采用 ICH E6(R2)在埃及开展临床试验的 GCP 的所有要素。埃及药品管理局关于临床试验行为的指导名为 “埃及药品管理局临床试验监管监督指南 ”。除 GCP 外,该指南还涵盖了埃及药品管理局特有的试验报告要求。
SAE在埃及要求迅速报告,因为 EDA 要求研究者在知晓任何致命或危及生命的 SAE 后 24 小时内 “通知 ”研究者,然后在通常的 7 天时限内完成 “个案安全报告”。此外,EDA 还要求 1 期和 2 期试验的 “非严重不良事件 ”必须在 7 天内提交给 EDA。指南中没有一处提到 15 天报告。指导原则中出现了 SUSAR 的定义,但没有明确指出只有 SUSAR 事件才需要按加速时限报告,因此,申办者应在试验授权过程中向 EDA 申请明确的加速报告要求。
该指南规定,在试验进行期间,应使用 EDA 的 “跟进模板 ”进行定期报告。对于持续时间少于一年的试验,这些报告应在试验开始后每三个月提交一次;对于持续时间超过一年的试验,应每六个月提交一次。
埃及没有针对特定国家的临床试验注册平台,但埃及药品管理局规定,凡获准在埃及开展的临床试验,必须在指定的三家注册机构中至少登记一家:FDA的 Clinicaltrials.gov、世界卫生组织的 ICTRP 或泛非临床试验注册平台 (PACTRP,Pan-African Clinical Trials Registry platform)。
TONACEA
黎巴嫩
黎巴嫩公共卫生部(Ministry of Public Health, MOPH)是黎巴嫩的药品监管机构。临床试验管理条例源自 “2014 年 6 月 23 日关于临床试验管理条例的第 1159/1 号部长令”,该法令为批准和开展研究性治疗药物的临床试验提供了法律和管理框架,也是在临床研究中保护人类受试者的法律框架。
在黎巴嫩,临床试验的审批程序是逐步进行的,申办者应直接向卫生部提交 CTA 档案:
• CTA 表
• 临床试验方案
• IB
提交 CTA 后,MOPH 将出具意见。规定的周转时间约为三周,但实际经验表明,试验审批流程第一步的周转时间差异很大。在收到澳门邮政储蓄银行的正面意见后,执委会将进行审查和批准。
与其他药品法律和监管框架更为健全的国家相比,黎巴嫩卫生部的临床试验法规要较短也不那么详细。不过,该框架中概述的试验行为和报告要求符合 ICH 原则,包括在 7 天(危及生命或致命)或 15 天(所有其他 USAR 事件)的时限内加快报告 “意外严重不良反应”。法令规定应使用 CIOMS I 表格(CIOMS I Form 在黎巴嫩临床试验中用于向监管机构快速、规范地报告个别受试者发生的严重、非预期不良反应(SUSAR))。黎巴嫩境外的申办者也可以使用 DSUR 格式进行年度安全报告,以取代 “年度进展报告 ”(yearly progress report)。
黎巴嫩有一个名为 “黎巴嫩临床试验登记处”(Lebanon Clinical Trials Registry,LCTR)的国家临床试验登记处,由于其健全的数据标准,符合纳入世卫组织 ICTRP 网络的要求。法律没有要求在黎巴嫩临床试验登记处登记试验,但黎巴嫩执委会可能会要求登记。
TONACEA
沙特阿拉伯王国
沙特阿拉伯食品药品管理局(Saudi Food and Drug Authority ,SFDA)是沙特阿拉伯王国卫生部(MoH)下属的政府机构,在法律上负责对治疗产品(包括药品和生物制品)进行监管。沙特是海合会(海湾合作委员会(GCC,简称“海合会”))的创始成员国之一,并与其他海合会成员国合作,以类似于欧洲药品管理局(EMA)集中授权程序的方式,建立了一个集中的医药产品注册程序。
SFDA的法定职权范围来自 2003 年 10 月 3 日(回历 1424 年 1 月 7 日)颁布的第 7/B/6629 号大臣会议皇家法令,该法令标志着国家食品药品监督管理局的成立,标志着国家食品药品监督管理局从卫生部接管治疗产品的监管工作。
SFDA 要求不在沙特阿拉伯王国( KSA(Kingdom of Saudi Arabia’s)) 境内的申办者通过境内 CRO 申请试验授权(学术或政府支持的研究可由 KSA 境内经批准的学术或医疗机构提交)。临床试验申请人必须首先使用沙特临床试验注册门户网站:沙特临床试验注册中心(Saudi Clinical Trials Registry,SCTR)进行注册。
与许多国家一样,沙特也有一个循序渐进的临床试验审批流程(先由地方监管部门审批,再由中央监管部门审批)。在注册之后,申办者可以先向相关的 IRB 申请伦理审批,然后再向 SFDA 临床试验管理局提交 CTA 文件包,因为提交 CTA 文件需要获得 IRB 的批准。国家医学与生物伦理委员会是沙特阿拉伯的主要伦理委员会。该委员会于 2001 年 8 月 8 日(回历 1422 年 5 月 18 日;农历)获得皇家法令批准,由阿卜杜勒-阿齐兹国王科技城直接管理。该委员会由分散在王国各地的多个地方伦理委员会组成,为参与治疗产品介入性临床试验的多家医疗机构提供服务。
获得 IRB 批准后,CTA 的两个主要途径如下:
- 在沙特阿拉伯注册的治疗产品试验;或
- 未在沙特阿拉伯注册的治疗产品试验、
以及
- 获得 FDA 和/或 EMA 批准的治疗产品;或
- 未经 FDA 和/或 EMA 批准的治疗产品。
在沙特进行的大多数试验属于第二类,即沙特或 FDA 或 EMA 均未注册的产品(许多 3 期研究在沙特进行),或 FDA 和/或 EMA 已批准但沙特未批准的产品(许多 4 期研究在沙特进行)。SFDA 对这些类型申请的 CTA 内容要求与其他 ICH 国家的要求非常相似,包括以下内容:
• 封面信(Cover letter)
• SCTR 注册
• 试验方案(Protocol)
• 知情同意书(阿拉伯语和英语)
• IRB/EC 批准
• IB
• 主要研究人员的财务披露(Financial disclosure of principal investigator)
• GMP 证书
• 研究药物分析证书(Certificate of analysis of study drug)
• 研究药物标签
• CTA
• 主要研究人员和协调员的简历(CV)
• 病例报告表(Case report form)
• 试验保险证明(Trial insurance certificate)
• 委托/授权书(针对 CRO)
CTA 的审查和批准过程相当漫长,大约需要 180 天,其中包括申办者解决疑问的时间。在 KSA 注册的产品的4期研究无需 SFDA 批准,只需试验申办者通知 SFDA 即可(Phase 4 studies of products registered in KSA do not require approval by SFDA and merely require notification to the SFDA by the trial sponsor.)。
截至 2020 年 11 月 30 日,SFDA已全面采用 ICH E6(R2) GCP,并将其纳入试验监管流程和指南。SFDA关于加快 SUSAR 报告的上市前药物警戒实践以及使用 DSUR 进行年度安全性报告的指导与相关 ICH 指导保持一致,并纳入SFDA的 “良好药物警戒实践指导原则 (GVP) ”。
在沙特阿拉伯(KSA)开展的所有临床试验——无论 I、II、III 期还是 IV 期——均必须在沙特临床试验注册系统(SCTR)上进行登记;沙特食品药品监督管理局(SFDA)明确要求在研究参与者入组之前完成登记。
TONACEA
非洲的临床试验监管
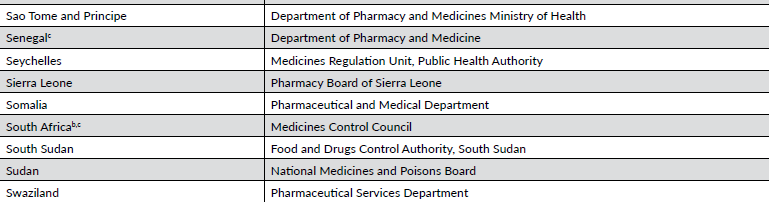
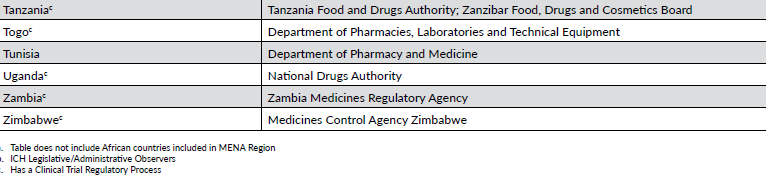
非洲是仅次于亚洲的第二大人口众多的地理区域 。尽管非洲人口占世界人口的很大一部分,但该地区的人口在临床试验中的代表性可能是最不足的。虽然大多数非洲国家都有一些国家级卫生机构负责医药产品的监管和监督(表 16-4),但只有一小部分国家(约 30%)制定了治疗产品注册和/或临床试验审批与实施的立法和监管程序。
在非洲大部分地区,建立必要的立法和监管基础设施,以承受临床试验监督和治疗产品注册的沉重负担,仍是一项进行中的工作。尽管如此,一些非北非和中东地区的非洲国家已经制定了强有力的现代化临床试验监督计划,其中包括南非、尼日利亚、喀麦隆和坦桑尼亚。
在 COVID-19 大流行期间,为开发新型 SARSCoV-2 疫苗而进行的全球临床试验凸显了非洲与世界其他地区在获得临床试验方面的差距。尽管该地区有几个国家具备临床试验能力,但在疫苗试验高峰期,只有两种 SARS-CoV-2 候选疫苗在非洲进行了试验,而且这些试验仅在南非进行。这个最新的例子突出表明,申办方必须增加在非洲国家进行试验的机会,并更好地利用那些已经建立了健全程序的非洲国家的试验监管和监督框架。
由于大多数非洲国家的临床试验基础设施有限,该地区在治疗产品开发、临床试验授权和执行实践方面是最不协调的地区之一。南非是非洲大陆唯一一个非中东和北非国家的 ICH 观察员。南部非洲发展共同体和东非共同体组织作为促进区域协调倡议组织的一部分被列入国际非物质文化遗产委员会成员,但目前尚不清楚这些组织在多大程度上能够使非洲的监管机构与国际非物质文化遗产委员会的指导方针保持一致。
非洲药品管理局(African Medicines Agency, AMA)是非洲一个相对新成立的多国组织,其成立的明确目的是统一整个非洲的治疗产品监管程序和指南,包括最终建立一个非洲新型治疗产品注册的集中程序。
非洲药品管理局是国际制药商协会联合会(International Federation of Pharmaceutical Manufacturers & Associations)的心血结晶,非洲联盟大会于 2019 年批准的一项条约促成了非洲药品管理局的成立。批准非洲药品管理局条约需要 15 个国家,但该组织正在积极努力,以尽快让 55 个非洲国家中尽可能多的国家参与进来。目前加入非洲医学协会的国家包括阿尔及利亚(中东和北非)、贝宁、乍得、刚果民主共和国、加纳、几内亚、马达加斯加、马里、摩洛哥(中东和北非)、尼日尔、卢旺达、阿拉伯撒哈拉共和国、塞内加尔、塞舌尔和突尼斯。
本节将重点介绍南非的治疗产品临床试验监督。
负责执行南非治疗产品注册和介入性临床试验相关法律法规的监管机构是南非保健品监管局(SAHPRA),这是一个独立/自治的监管实体,通过成立的委员会向国家卫生部报告。 南非成立并授权南非保健品监管局的主要治疗监督法规是《药品及相关物质法》(MRSA;1965 年第 101 号法案)。MRSA 第 21 条专门涉及有关介入性临床试验的法律法规。此外,SAHPRA 的法律授权来自 1996 年《南非共和国宪法》和 2003 年《国家卫生法》(2003 年第 61 号法案)。
介入性临床试验法规由 南非保健品监管局(SAHPRA)内的临床试验部门负责管理。临床试验部门在 SAHPRA 网站的 “临床试验 ”业务单元下列出了所有 CTA 表格和指南: 申办者首先必须获得必要的欧盟委员会批准,然后填写 CTA 表格并提交以下文件:
• 封面信(PDF 和 MSWORD 格式的签名副本各一份)
• 两份填写完整的 CTA(一份以 PDF 格式签署,一份以 MS-WORD 格式签署)
• 临床研究方案
• 患者信息宣传单和知情同意书
• 招聘广告(如适用)和调查问卷
• IB 和/或所有专业信息[包装插页]
• 分析证书
• SAHPRA格式的研究者简历(在CTA表格附录中提供
• 所有研究者的声明(在 CTA 表附录中提供
• 经签署的联合财务声明(申办者和国家研究员;在 CTA 表附录中提供)
• 申请人和国家首席研究员签署的声明(在 CTA 表附录中提供)
• 地区监查员的简历和签名声明
• 申请在南非国家临床试验登记簿上登记试验的证明
• 有效的临床试验保险证明
• 申请人对研究人员和试验场地的赔偿证明
• 有效的 GCP 证书
• 研究者工作量表(在 CTA 表附录中提供)
• 目前在专业法定机构的注册证明
• 当前的专业赔偿(失职保险)证明
• 伦理批准书或提交给伦理委员会的批准书副本
• 研究预算
• 参考文献
• 研究产品生产的 GMP 证书
• 指定实验室的认证/证书证明
CTA申请通过电子邮件(和上述硬拷贝)提交至SAHPRA,并遵循SAHPRA新临床试验流程/预批准流程,其中包括由专门组建的专家委员会成员对申请进行审查。整个过程大约需要 60 到 90 天。在提交 CTA 之前,试验必须在南非国家临床试验登记簿上登记。
SAHPRA 网站的 “临床试验指南 ”部分在线发布了 11 项临床试验指南。这些指南包括但不限于《SAHPRA GCP 指南》, 该指南与 ICH E6(R2)、《临床试验的监督与监测》和《临床试验期间的安全性报告》 相一致。SAHPRA 还专门制定了旨在提高南非临床试验机构临床试验能力的指导原则,其中包括一项规定:“如果一项试验涉及四个或更多试验机构(多中心临床试验),则至少有 25% 的试验机构应为公共机构和/或培训机构,除非有明确的理由说明为何不能这样做”。
SAHPRA临床试验药物警戒定义和快速事件(SUSAR)报告与ICH完全一致,DSUR格式可作为年度试验更新报告的一种方式。
SAHPRA 管理着一个国家临床试验登记处--南非国家临床试验登记处,并依法要求所有临床试验在启动前必须进行登记。
在全球范围内,由于 ICH、CIOMS(Council for International Organizations of Medical Sciences) 和 WHO 等跨国组织的努力,对临床试验审批和实施的监管正朝着更加协调的方向发展。这些组织及其他类似组织为药品监管机构、区域协调组织(如亚太经合组织、IPRP)和业界提供了一个合作框架,以制定临床试验监管的最佳实践。
自 20 世纪 90 年代以来,经济全球化迅速发展,导致世界上药物开发历来滞后的地区对新型疗法的需求增加,从而促使亚太和中东及北非等地区加大了临床试验协调和准入力度。为实现临床试验监管的区域集中化和标准化所做的努力,包括欧盟更新的《临床试验条例》[Regulation (EU) No 536/2014]等已实现的努力,以及非洲药品管理局和海湾合作委员会等处于早期发展阶段的努力,都有助于加快临床试验监管协调的时间框架。
除了统一药物集中监管机构的做法外,继续统一地方 IRB/REC 审批、改善与国家卫生研究所和美国癌症协会等资助机构的合作、开发申办方模式以适应标准化的保险和赔偿,这些都是可以长期实现的目标。
继续努力最大限度地减轻研究参与者的临床试验负担,降低参与研究的障碍(例如,周到地纳入 DCT 要素并加强对其使用的监管指导),以及切实努力提高临床试验的多样性(例如,FDA 的 “提高临床试验中代表性不足的种族和民族参与者的多样性计划”),对于临床开发计划的发展至关重要。这些努力将有助于为未来建立一个全球性的基础设施,以便高效、有效地开展跨国试验。与其他国家的政府和监管机构建立合作关系,促进科学和临床治疗的发展也很重要。
建立一套通用的全球数据要求将提供标准化的信息,从而节省时间、金钱,并最终挽救生命。这些监管流程应能促进稳健而及时的临床开发,而不是成为限制创新潜力、造福人类的障碍;这才符合人们的整体利益。

上一页
相关新闻

联系我们
同写意

写意宣发

同写意Biotech

同写意微服务

©2022 同写意(北京)科技发展有限公司