
辉瑞54亿美元押注折戟
更新时间:
2025-08-17 18:17:59.541

这已是辉瑞在SCD战场遭遇的“三连败”:
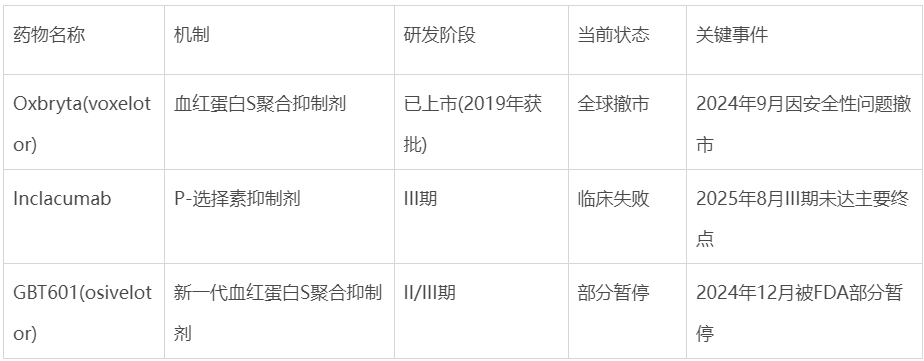
• 9个月前,拳头产品Oxbryta因“死亡人数异常增加”遭全球撤市,欧洲药管局报告显示用药组死亡率超安慰剂组3倍;
• 5个月前,被寄予厚望的下一代口服药GBT601遭FDA全面叫停试验;
• 此刻,最后一款主力在研药物倒在临床终点线前。
时间回溯至2022年8月,辉瑞以54亿美元高价收购专注于SCD治疗的生物技术公司GBT,这笔交易被视为辉瑞进军遗传性血液疾病领域的关键布局。通过此次收购,辉瑞获得了三款核心资产:已上市药物Oxbryta(voxelotor)、临床后期候选药物inclacumab以及下一代血红蛋白聚合抑制剂GBT60110。
彼时,SVB证券分析师曾乐观预测,GBT的管线资产尤其是GBT601作为“功能性治愈”疗法的潜力,足以为这一收购价格提供充分支撑。如今54亿美元换来的,只有冰冷的临床数据与破碎的30亿美元年销售幻梦。
TONACEA
挫败的辉瑞
当辉瑞在2022年豪掷巨资收购Global Blood Therapeutics时,它赌的是SCD领域“小分子药物+基因疗法”的双轨制未来。但现实是,传统药物开发逻辑正在基因编辑革命前土崩瓦解——就在Oxbryta撤市当月,两款天价基因疗法Casgevy与Lyfgenia获FDA批准上市,220万美元的单次治疗要价,宣告“一次性治愈”时代的降临。
这一系列挫折背后,辉瑞最初对SCD领域的战略预判与商业野心值得深究。
在收购GBT时,辉瑞血液学研发重点本就聚焦于SCD和血友病领域,与GBT的专长高度契合。通过整合Oxbryta及两款在研药物,辉瑞有望在SCD领域实现年销售额超过30亿美元的业绩。然而现实残酷——2023年Oxbryta全球销售额仅3.28亿美元,2024年第二季度更降至9200万美元,远不及预期。
更令辉瑞陷入被动的是,inclacumab曾被视为解决SCD治疗痛点的潜力产品。作为一种靶向P-选择素的全人源单抗,inclacumab通过选择性阻断P-选择素蛋白发挥作用,可减少血管阻塞危象的发生并减轻疼痛症状。其最大优势在于每季度给药一次的便利性,显著优于当时需每月输注的竞品。
2022年6月,该药凭借“同类最佳”潜力获得FDA的孤儿药和罕见儿科疾病认定。然而,患者招募困难已初显端倪——2024年3月,辉瑞因患者招募缓慢(仅完成280名计划中的78名)终止了一项inclacumab的III期研究。
辉瑞在SCD领域的困局不仅是一次收购的得失,更揭示了罕见病药物开发中高风险与高回报并存的残酷现实。当54亿美元押注遭遇临床失败连环冲击,辉瑞不得不重新审视其在整个SCD治疗领域的战略定位与资源配置。
而这一系列挫折恰逢基因疗法在该领域取得突破性进展,传统小分子药物开发模式面临前所未有的挑战。
TONACEA
双重困境
镰状细胞病作为一种遗传性血液疾病,其病理机制已相对明确:患者血红蛋白β链基因突变导致血红蛋白S(HbS)异常聚合,进而使红细胞变形为镰刀状。这些异常细胞不仅携氧能力低下,还会阻塞微血管,引发血管闭塞危象(VOC)、慢性溶血性贫血、多器官损伤等一系列临床表现。全
球每年约有500万人死于该疾病,在非洲、中东、印度等地肆虐。值得注意的是,随着全球人口流动,SCD在欧美国家的发病率持续上升——美国疾病控制与预防中心(CDC)数据显示,SCD在美国人群中的发病率已达1/3300,患者超过10万人,主要为非裔美国人;在法国,SCD甚至以1/2415的新生儿发病率超越其他遗传病,成为该国最常见的遗传病。
长期以来,SCD治疗领域面临选择有限、副作用大、疗效欠佳的困境。
• 羟基脲(Hydroxyurea):作为最早获批的SCD治疗药物,虽能一定程度上诱导胎儿血红蛋白表达,但其骨髓抑制等严重副作用限制了广泛应用。
• Endari(L-谷氨酰胺):2017年获FDA批准,成为20年来首个SCD新药,但临床效果有限。
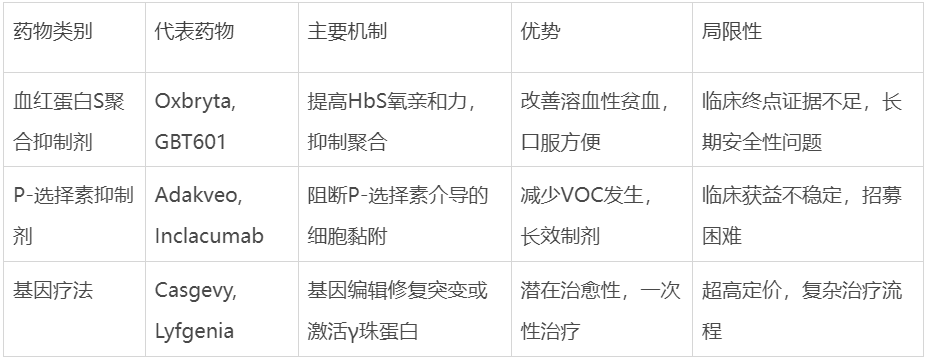
• Adakveo(crizanlizumab):诺华2019年推出的P-选择素抑制剂,作为首个靶向治疗SCD的药物曾备受期待,2022年全球销售额近2亿美元。然而2023年1月,该药在III期研究中未能降低VOC发生率,前景蒙尘。
• Oxbryta(voxelotor):由GBT研发,2019年获FDA加速批准,作为全球首款血红蛋白S聚合抑制剂,通过增加血红蛋白对氧的亲和力,阻断镰状化过程。该药曾获“医药界诺贝尔奖”盖伦奖,2023年全球销售额3.28亿美元。然而其安全性问题最终导致撤市。
这些传统药物共同面临的核心挑战在于:仅能部分缓解症状,无法解决疾病根本病因,且长期使用可能带来难以预料的安全性问题。
Oxbryta的撤市更是凸显了这类药物在长期安全性评估中的不确定性——尽管在加速审批时显示出改善贫血的效果,但在更大规模、更长周期的研究中,其血管闭塞危象和致命事件风险才逐渐显现2。
辉瑞从GBT获得的药物管线,其科学基础主要围绕两个作用机制:
血红蛋白S聚合抑制(Oxbryta和GBT601):通过提高HbS与氧的亲和力,防止红细胞镰状化;P-选择素阻断(inclacumab):通过抑制介导细胞黏附的P-选择素,减少血管炎症和VOC发生。
然而,THRIVE-131研究的失败揭示了这些机制的局限性。inclacumab虽然在II期研究中显示出减少VOC的潜力,但在更严格的III期随机对照试验中,这一效果未能转化为统计学显著获益。同样值得注意的是,GBT601在2023年美国血液学会(ASH)年会上公布的II/III期初步数据显示,该药能显著提高血红蛋白水平(150mg剂量组平均提高3.17g/dL),且改善红细胞变形能力。
但血红蛋白水平升高这一替代终点是否真能转化为VOC减少的临床终点,始终存在疑问——辉瑞最近的挫折似乎给出了否定答案。
SCD药物开发的另一重困境在于商业回报与患者分布的错位。该疾病主要影响撒哈拉以南非洲地区人群,而这些地区的支付能力极为有限。即使在发达国家,SCD患者也多为社会经济地位较低的少数族裔。这种分布特点导致制药公司在定价与市场预期间陷入两难。
GlobalData曾预测,到2030年美国镰状细胞疗法市场规模约48亿美元;Fortune Business Insights则估计全球市场可达98.4亿美元。然而现实是,即使是已上市的创新药物,也面临市场渗透不足的挑战。Oxbryta在2023年全球销售额仅3.28亿美元,远低于预期。而基因疗法的天价定价(220万-310万美元)更是将绝大多数患者拒之门外。
辉瑞54亿美元押注的连续失败,不仅是一家公司的挫折,更凸显了整个SCD小分子药物开发领域面临的科学不确定性与商业可持续性挑战。当传统药物开发路径遭遇瓶颈,基因疗法的崛起正在重塑整个治疗格局。
TONACEA
基因疗法是解药?
在传统小分子药物屡遭挫折之际,基因疗法为镰状细胞病治疗带来了革命性突破。
2023年12月8日,美国FDA同时批准了两款治疗SCD的基因疗法——Vertex/CRISPR Therapeutics的Casgevy(exagamglogene autotemcel)和蓝鸟生物的Lyfgenia(lovotibeglogene autotemcel),标志着基于基因编辑的SCD治疗正式进入临床应用阶段。
这两款基因疗法虽然针对相同适应症(12岁及以上复发性VOC的SCD患者),但采用了截然不同的技术路径。
• Casgevy:基于CRISPR/Cas9基因编辑技术,通过在患者自体造血干细胞中编辑BCL11A基因,重新激活γ-珠蛋白表达,增加具有抗镰状化能力的胎儿血红蛋白(HbF)5。在关键临床试验中,Casgevy展现出卓越疗效——超过90%的患者在18个月内未经历VOC,摆脱了痛苦的血管阻塞危象。
• Lyfgenia:采用慢病毒载体技术,将经过工程改造、编码具有抗聚合特性血红蛋白的基因导入患者造血干细胞,使红细胞产生HbAT87Q——一种能够抑制镰状血红蛋白聚合的功能性血红蛋白。
这两款疗法的获批不仅为SCD患者提供了潜在治愈性选择,更代表了医学史上的里程碑——Casgevy成为全球首个基于CRISPR技术的基因编辑疗法,开启了精准基因治疗新时代。然而,基因疗法的临床应用仍面临多重挑战。
尽管疗效显著,基因疗法的临床应用却进展缓慢。
Casgevy定价220万美元,Lyfgenia更高达310万美元。如此高昂的价格远超常规药物,即使在美国医疗保障体系中也面临支付障碍。基因疗法需要采集患者自体造血干细胞,在专业GMP设施中进行基因修饰,再回输给经过清髓预处理的患者。整个过程耗时数月,且清髓化疗伴随感染、不孕甚至继发癌症等风险。
目前全球仅有少数医疗中心具备实施基因治疗的能力,导致患者地理可及性严重受限。
据悉,两款基因疗法获批后数月内,仅有几十名患者接受了治疗。这种缓慢的商业化进程反映了超高值基因疗法在现实世界中面临的困境——即使科学成功,商业转化仍障碍重重。
为突破当前基因疗法的局限,全球研究团队正积极探索下一代技术。
• 体内基因编辑(in vivo gene editing):避免复杂的外周干细胞采集、体外编辑和回输过程,直接在患者体内靶向造血干细胞进行基因编辑。
• 非病毒递送系统:尧唐生物的技术采用优化脂质纳米颗粒而非病毒载体,降低了免疫原性和生产成本。在临床前研究中,该系统实现了高效体内基因编辑,且未诱发强烈免疫反应或器官损伤,全基因组范围内未检测到明显脱靶。
• 无需清髓预处理:传统干细胞基因治疗需要清空骨髓腔以接纳改造后的干细胞。尧唐生物的技术通过体内精准靶向造血干细胞,避免了清髓化疗的毒副作用,大幅降低治疗风险。
这些创新技术有望解决当前基因疗法的核心痛点——复杂的治疗流程、高昂的生产成本和清髓预处理的毒性,为基因治疗提供更安全、可及性更高的方向。
TONACEA
中国力量
当辉瑞等跨国药企在镰状细胞病治疗领域遭遇挫折时,中国生物科技公司正以创新技术重塑竞争格局。
2025年8月,上海尧唐生物在Nature Biomedical Engineering发表的重磅研究,为全球SCD治疗带来突破性进展——首次实现体内造血干细胞基因编辑,为“无需清髓即可一次治愈”提供了科学基础。
尧唐生物团队开发的LNP-mRNA疗法,直击当前SCD基因治疗的三大痛点:
• 复杂流程简化:传统基因疗法需采集患者干细胞、体外编辑、扩增后回输,全程耗时数周至数月。而尧唐生物的新型脂质纳米颗粒LNP-168,通过静脉注射即可在体内靶向造血干细胞,实现基因编辑。该技术无需干细胞采集与体外操作,大幅简化治疗流程。
• 清髓预处理消除:为避免患者自身未编辑干细胞竞争,传统方法需使用高剂量化疗药物清除骨髓。尧唐生物的技术通过优化LNP递送效率,实现了对造血干细胞的高效靶向,在动物模型中未进行清髓预处理即实现持久编辑效果。
• 生产成本降低:与病毒载体相比,LNP-mRNA系统的生产工艺更简单、成本更低,且免疫原性风险更低。这为基因治疗的可及性提供了重要保障。
在机制上,研究团队通过筛选可电离脂质库,开发出无需抗体修饰即可靶向骨髓的LNP-168。该载体可将腺嘌呤碱基编辑器(ABE8e)mRNA和靶向γ-珠蛋白(HBG1/2)启动子的sgRNA递送至造血干细胞,高效编辑HBG1/2启动子,重新激活胎儿血红蛋白表达。
在β-地中海贫血患者来源的HSC移植小鼠模型中,单次注射即可显著提高红系细胞胎儿血红蛋白含量,改善病理表型,且未检测到明显脱靶效应或器官损伤。
尧唐生物的技术并非停留在实验室阶段,其体内基因编辑平台已进入临床转化,首个适应症针对转甲状腺素蛋白淀粉样变性(ATTR)的体内基因编辑药物YOLT-101已于2024年3月获中国CDE批准临床试验,2024年6月完成首例患者入组,2025年1月进入剂量拓展阶段。
辉瑞的挫折警示我们,单一技术路径或传统商业模式在复杂罕见病领域风险极高。未来成功的企业将是那些能够整合科学创新、商业智慧和全球健康视野的整合者。
当基因编辑技术以惊人速度发展,治疗可及性不再应是科学突破后的补充考量,而应成为研发初期的核心设计要素。
SCD治疗的未来不仅关乎科学突破,更关乎对医疗公平的重新定义。在这个意义上,辉瑞54亿美元押注的教训和中国体内基因编辑的突破,共同构成了医疗创新史上的关键时刻——当技术能力与人文关怀真正融合,医学才能实现其最崇高的承诺:为所有患者提供希望,无论其出身何处、财富多寡。
上一页
相关新闻

联系我们
同写意

写意宣发

同写意Biotech

同写意微服务

©2022 同写意(北京)科技发展有限公司